阿司匹林是有上百年历史的消炎镇痛药;青霉素是有效治疗多种细菌感染的第一批抗生素;亿珂(伊布替尼,英文商品名Imbruvica)是改变血液癌症治疗的重磅药物。Lumakras是击破KRAS不可成药性的里程碑。这些在人类医药史上出现的重要药物都有一个共同点,它们都属于共价药物(covalent drug)。它们能够与靶点形成共价键,从而不可逆地改变靶点的功能。
然而很久以来,药物研发人员在进行新药开发时会有意避免与靶点共价结合的化合物。他们担心这些化合物的选择性不够强,一旦与不相关的蛋白结合,就可能产生严重的毒性。过去30年来,理性设计共价药物的开发理念逐渐受到人们的关注,它们不但产生了有效的重磅抗癌疗法,而且为靶向不可成药靶点提供了新策略。近日,Nature Reviews Drug Discovery的一篇综述对共价药物开发的最新进展进行了盘点。
人类使用共价药物的历史已经有上百年。阿司匹林最初在1899年就被使用作为消炎和镇痛药。不过直到1971年,人们才了解它的作用机制。阿司匹林能够与负责合成前列腺素和血栓素的环氧化酶(cyclooxygenase,COX)不可逆结合,从而抑制COX的活性,降低炎症的产生。▲阿司匹林的分子结构式(图片来源:public domain)
目前大多数小分子药物属于非共价抑制剂,它们与靶点的结合是可逆的,与靶点的亲和力往往决定了药物的治疗效果,因此大多数小分子药物的研发目标是发现与靶点以高亲和力结合的化合物。共价药物与靶点结合之后,化合物上的活性基团能与靶点产生化学反应形成共价键,这让共价药物与靶点蛋白形成一个稳定的复合体,从而不可逆地抑制靶点蛋白的活性。这种作用方式具有独特的优点,不可逆地抑制靶点蛋白的活性意味着在细胞生成新的靶点蛋白之前,共价药物都能维持疗效。这可能导致共价药物只需要更少的剂量和服药次数,就能达到与非共价药物相同的疗效。而且研究显示,共价药物与非共价药物相比,在靶向容易产生耐药性的靶点方面更具优势。▲共价药物(b)和非共价药物(a)的作用方式(图片来源:参考资料[5])然而,共价抑制剂让人们担忧的因素是它的潜在毒副作用。因为这些化合物能够与蛋白形成共价键,与非靶点蛋白的结合可能造成严重的副作用。2013年,FDA批准第二代EGFR抑制剂阿法替尼(afatinib,英文商品名Gilotrif)和布鲁顿氏酪氨酸激酶(BTK)抑制剂伊布替尼(ibrutinib)上市。与此前获批的共价药物不同,它们是首批有意设计的共价抑制剂。研究人员将能够与靶点形成共价键的活性基团添加在已有的可逆抑制剂的骨架上,有效提高了药物对靶点的活性。阿法替尼能够持久地抑制EGFR活性并且部分克服导致可逆性EGFR抑制剂失效的T790M突变。而伊布替尼不但能够高效和持久地抑制BTK活性,它能够被身体迅速清除的特征也限制了潜在的毒副作用。在2013年获批治疗套细胞淋巴瘤后,伊布替尼迅速获得FDA批准治疗包括慢性淋巴细胞白血病和华氏巨球蛋白血症(Waldenstrom macroglobulinemia)在内的多项适应症,被人们称为让患者脱离化疗的重磅新药(关于这款新药开发的详细历史,请看亿珂诞生记:让化疗成为历史的重磅新药)。日前,它斩获了第12项FDA批准。伊布替尼的成功证明理性设计的共价药物不但可以获得可以接受的安全性,而且具有成为重磅药物的潜力。它与一系列EGFR共价抑制剂的设计也体现了共价药物开发的一大策略:基于与靶点可逆性结合的化合物,在它们的骨架上添加与靶点形成共价键的化学基团。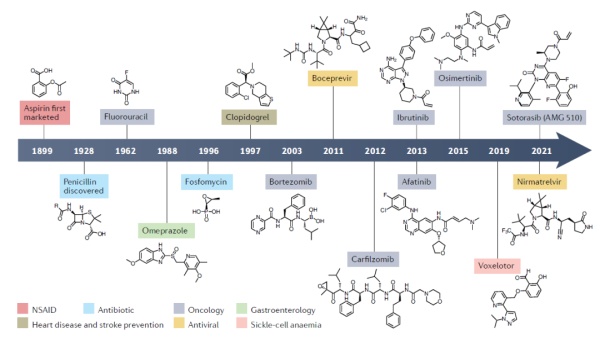
▲历史上获批的部分重要共价药物(图片来源:参考资料[1])
共价药物开发的另一种策略从与靶点形成共价键的亲电基团入手,在化合物发现过程中首先专注于发现能够与靶点共价结合的配体。这一策略的代表是KRAS G12C共价抑制剂。传统的非共价药物需要与靶点以高亲和力结合才能发挥作用,而很多靶点蛋白的表面缺乏能够让小分子有效结合的“口袋”,这让它们很难被非共价药物靶向,KRAS就是其中之一。虽然它和RAS家族其它成员的基因突变在30%的癌症中出现,但是几十年来研发人员一直无法找到合适的靶向药物。在2013年,加州大学旧金山分校的Kevan Shokat教授率领的团队发现,在名为KRAS G12C的突变体中,因为基因突变生成的半胱氨酸(Cys)可以与小分子的亲电基团生成共价键。由于KRAS G12C突变只出现在肿瘤细胞中,靶向这一突变体不会影响到健康细胞中的野生型KRAS。针对KRAS G12C突变体上的半胱氨酸,研究人员开发出能够与之共价结合的小分子抑制剂。这一发现激发了多家公司开发靶向KRAS G12C的共价抑制剂。其中,安进(Amgen)公司的Lumakras在去年获得FDA加速批准,成为首款获批的KRAS靶向疗法。Mirati Therapeutics公司的adagrasib也正在接受美国FDA的审评,有望在今年获得批准。KRAS G12C共价抑制剂的成功也代表着在共价药物开发理念上的一个转变。此前,共价药物的开发通常基于对非共价药物的改造,而KRAS G12C共价抑制剂的发现策略则是直接筛选能够与突变体上的半胱氨酸生成共价键的候选小分子化合物。这种筛选方式虽然没有发现与突变体以高亲和力结合的化合物,但是发现了与半胱氨酸快速生成共价键的化合物。它们的成功显示,对于共价抑制剂来说,只要接触到靶点的分子能快速生成共价键,就可以达到抑制靶点活性的作用。这为药物开发人员提供了寻找候选化合物的一个新方向。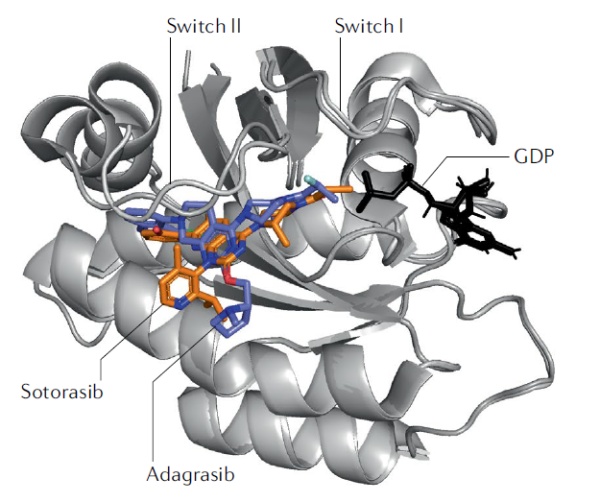
▲Lumakras和adagrasib与KRAS G12C突变体结合的结构示意图(图片来源:参考资料[1])
针对KRAS突变体上出现的半胱氨酸设计共价药物的成功也意味着在其它疾病中,如果基因突变产生半胱氨酸,共价抑制剂是开发潜在精准疗法的有力策略。这一策略的新近例证是辉瑞公司开发的COVID-19口服疗法Paxlovid。它的主要活性成分nirmatrelvir通过与新冠病毒Mpro蛋白酶上的半胱氨酸共价结合,达到抑制病毒复制的作用。如今,理性设计共价药物来靶向“不可成药”靶点已经成为多家公司小分子药物开发工具箱中的重要工具。新近的多项筛选技术能够让研发人员直接筛选与靶点共价结合的化合物片段。DNA编码化合物库(DEL)的出现也进一步提高了科学家们可以筛选的化合物数量。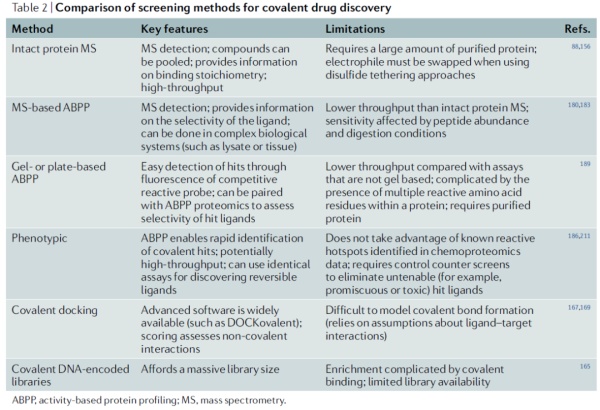
▲共价药物发现筛选策略的比较(图片来源:参考资料[1])
化学蛋白组学(chemoproteomic)平台的进步也让研发人员能够有效地衡量共价药物的特异性,从而减轻了对它们毒副作用的疑虑。这一技术以可形成共价键的化合物为“饵”,对整个蛋白质组进行筛选,找出能与化合物结合的蛋白片段。它不但能够检测出共价药物的特异性,而且可能帮助筛选出全新的成药位点。综述的作者预计,未来的共价药物开发将更多使用直接筛选与靶点结合的共价配体的发现方式,尤其是在与靶点可逆性结合的化合物难于发现的情况下。此外,对可逆性共价机制的探索有望进一步增多。这一机制在多种情况下可能帮助找到疗效和选择性之间的平衡。鉴于共价药物开发在克服不可成药靶点方面的成功,它将成为更好发挥小分子药物潜力的重要工具。



{replyUser1} 回复 {replyUser2}:{content}