《Science》封面:用SMRT长读长测序技术挖掘人与猿真正差异
长期以来,我们都在努力的去研究与发现是怎样的遗传差异造就了我们人类,对猿谱系内所有形式的遗传变异进行全面的发现与比较,则是完成这一研究不可或缺的方法。
——Kronenberg et al., Science, 8 June 2018

2018年6月8日,美国华盛顿大学著名的遗传学家Evan Eichler及其团队发表了Science封面文章,题为“High-resolution comparative analysis of great ape genomes”,该论文运用SMRT长读长测序技术,Bionano光学图谱技术,完成并组装了黑猩猩(Chimpanzee),猩猩(Orangutan)和两个人(Human)的基因组,然后将它们与先前的大猩猩(Gorilla)基因组进行了深度的比较。
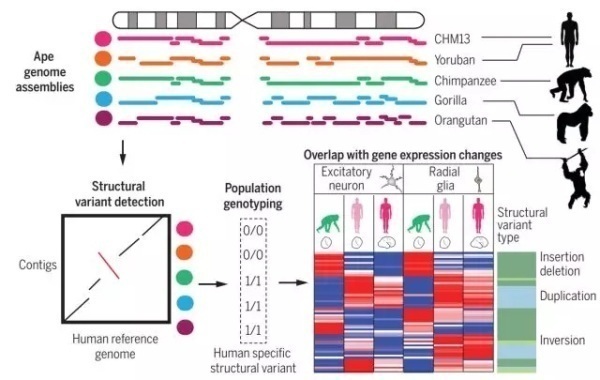
图1:SMRT测序组装及SV分析
在以往猿类的研究中,基因组组装往往存在上千个gap,并且在组装过程中使用了人类基因组作为参考,使得猿类的基因组在一开始就已经具备了“人源性”。不仅如此,这也使得在基因组层面,相当多的结构变异(Structural Variant,SV)及其他类型的变异被掩盖,并且也限制了人类与其他猿类的功能差异的比较与发现。
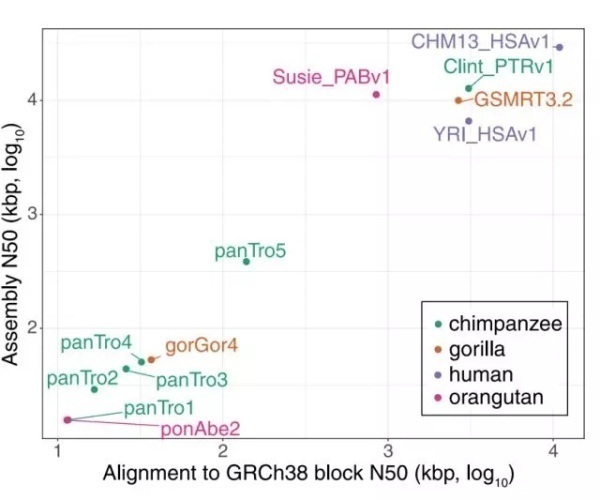
图2:先前的参考基因组(ponAbe2,gorGor4,panTro2,panTro3,panTro4,panTro5)与SMRT测序技术得到的基因组(黑猩猩Clint_PTRv1,猩猩Susie_PABv1,大猩猩GSMRT3.2,人类CHM13_HSAv1和YRI_HSAv1)质量比较
Eichler在报告中指出,“我们的目标是得到多种与人类基因组一样高质量的猿基因组,只有这样才能使我们真正了解哪些基因差异使我们成为了独一无二的人类。”
为了解决这些问题,Eichler带领了来自十多个机构的40多位研究人员组成的团队,运用长读长的SMRT测序技术,Bionano光学图谱技术对两个人,一个黑猩猩和一个猩猩基因组进行了深度超过65X的de novo测序。大约93%的黑猩猩和猩猩的碱基序列能够在没有人类参考基因组作为辅助,不引入人源性偏差的情况下,组装成为染色体水平的scaffold。其中存在于先前的黑猩猩的基因组中的7,797个gap,在这项研究中已有52%被填补完成;在猩猩常染色体基因组中的315,124个gap已成功填补了96.8%(305,069个);这与以前的工作相比,基因组质量提高了32~533倍。然后,将基因组组装结果进行比较,进而梳理出了特异性差异。
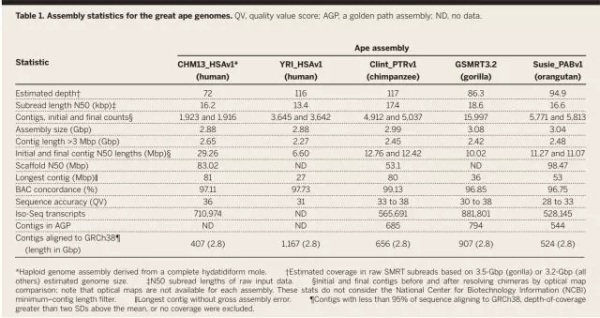 表1:长读长测序技术获得的五个高质量基因组总览,其中Contig N50分别为:人类29.26Mb及6.60Mb,黑猩猩12.76Mb和12.42Mb,大猩猩10.02Mb;猩猩11.27Mb和11.07Mb;其中,经过Bionano光学图谱技术进行黄金组装提升(A Golden Path Assembly,APG),黑猩猩,猩猩和大猩猩中,contig的数量分别减少至685个,794个以及544个
表1:长读长测序技术获得的五个高质量基因组总览,其中Contig N50分别为:人类29.26Mb及6.60Mb,黑猩猩12.76Mb和12.42Mb,大猩猩10.02Mb;猩猩11.27Mb和11.07Mb;其中,经过Bionano光学图谱技术进行黄金组装提升(A Golden Path Assembly,APG),黑猩猩,猩猩和大猩猩中,contig的数量分别减少至685个,794个以及544个
研究人员们还利用了Iso-Seq技术,对来自黑猩猩、猩猩、以及大猩猩的多能干细胞的转录组水平的研究,进而开发新的基因模型。
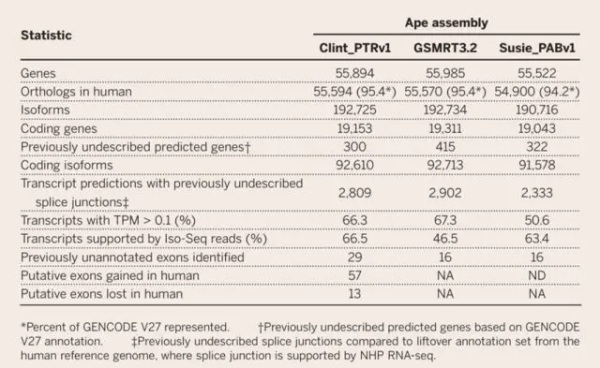 表2:猿类基因及转录本注释,分别在黑猩猩(Clint_PTRv1),大猩猩(GSMRT3.2)和猩猩(Susie_PABv1)中鉴定到的基因数为192,725个,192,734个和190,716个;其中,分别有300个,415个 及322个基因在之前的描述中未被预测到
表2:猿类基因及转录本注释,分别在黑猩猩(Clint_PTRv1),大猩猩(GSMRT3.2)和猩猩(Susie_PABv1)中鉴定到的基因数为192,725个,192,734个和190,716个;其中,分别有300个,415个 及322个基因在之前的描述中未被预测到
在对人类,黑猩猩,猩猩以及之前得到的大猩猩的五种全基因组的序列比对中,研究人员发现,83%的猿基因组可用于比较。
在基因组的深入分析研究中,研究人员们发现,重复元件往往出现组装的错误。他们发现黑猩猩和大猩猩中存在内源性逆转录病毒PtERV1,而在猩猩和人类基因组中则并没有。随后,他们定位了“源PtERV1”——一种直系同源黑猩猩和大猩猩PtERV1的元件。 这一元件由于不完整的谱系选择,可能没有在人类谱系中得以发现。但研究人员们还提到,由于这是一个位于重复序列富集区域中的元件,因此在之前的研究中没有被发现。
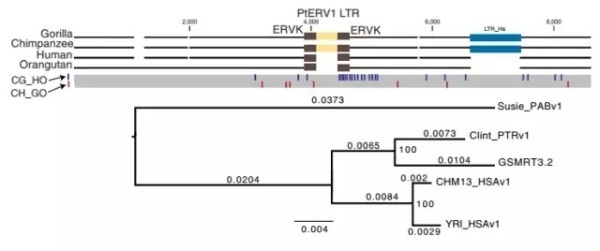 图3:体现大猩猩与黑猩猩直系同源的PtERV1元件,在人类以及猩猩中则并未出现。
图3:体现大猩猩与黑猩猩直系同源的PtERV1元件,在人类以及猩猩中则并未出现。
研究人员们还鉴定到了了猿谱系中超过614,186个的缺失,插入和倒位变异事件。与此同时,他们发现了17,789种固定的人类特定结构变体(fixed human-specific structural variant,fhSV)。对这些相对于黑猩猩和人类基因模型的fhSV进行注释,研究人员们还发现,这些变异影响了将近650个调控区域的480个基因。
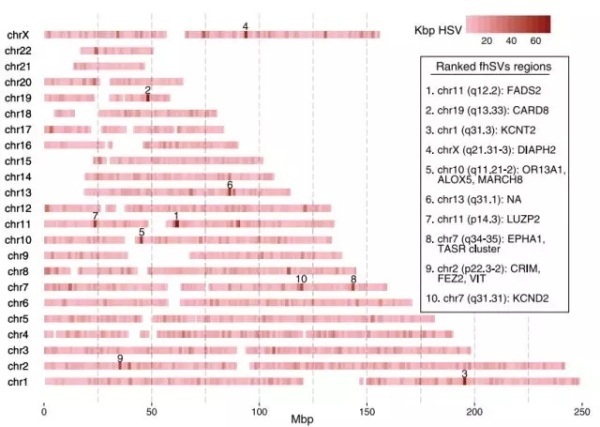 图4 fhSV图谱,颜色越深代表SV涉及的碱基数越多
图4 fhSV图谱,颜色越深代表SV涉及的碱基数越多
例如,一个fhSV,为参与脂肪酸生物合成的FADs1和FADs2基因出现了部分的缺失。而另一些fhSV影响了细胞周期基因WEE1和CDC25C,从而导致额外的细胞分裂。研究人员指出,额外的细胞分裂被认为与人类皮质神经元数量的增加有关。而总体上看,这些变化则可能对猿类和人类之间的一些差异进行解释。
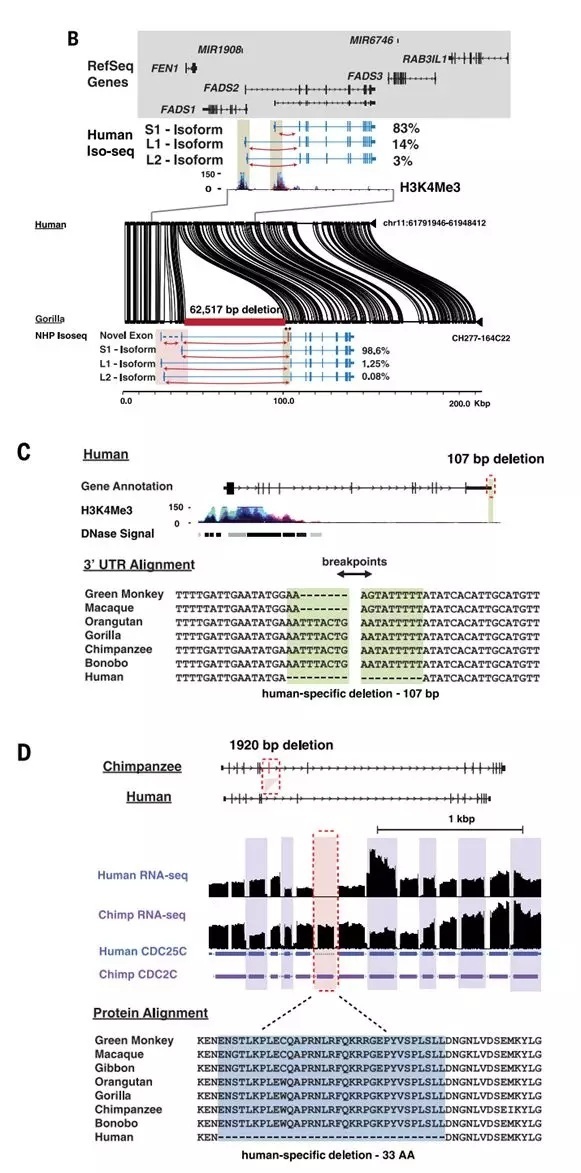 图5:B,FADS2基因中62.5kb的缺失;C,WEE1基因中,3′UTR端107bp的缺失;D,细胞周期调控相关基因CDC25C中1,920bp的缺失
图5:B,FADS2基因中62.5kb的缺失;C,WEE1基因中,3′UTR端107bp的缺失;D,细胞周期调控相关基因CDC25C中1,920bp的缺失
而在研究过程中,面对一些更大更复杂的基因组结构变异,作者采用了结合Bionano光学图谱的技术。进而得到了准确的结构变异信息。
 图6:A,运用Bionano光学图谱技术,在黑猩猩中检测到的染色体13q14.3上约为265kb的倒位;B,位于黑猩猩染色体13q14.13上,通过Bionano光学图谱检测到的约为1.1Mb的倒位变异,包含了15个基因;C,通过Bionano光学图谱和BAC末端测序检测到的染色体倒位,后续通过分裂中期分析,以及分裂间期的FISH实验得以证实。
图6:A,运用Bionano光学图谱技术,在黑猩猩中检测到的染色体13q14.3上约为265kb的倒位;B,位于黑猩猩染色体13q14.13上,通过Bionano光学图谱检测到的约为1.1Mb的倒位变异,包含了15个基因;C,通过Bionano光学图谱和BAC末端测序检测到的染色体倒位,后续通过分裂中期分析,以及分裂间期的FISH实验得以证实。
这篇文章通过SMRT长读长测序技术,Bionano光学图谱技术对两个人类,一个黑猩猩和一个猩猩基因组进行了测序,在没有使用人类基因组作为指导的条件下,更为客观的将我们的近亲的基因组置于与人类基因组相同的条件下进行比较。同时还运用了Iso-Seq方法对超过500,000个RNA分子进行测序,从头开始构建基因模型,从而增加了我们对每个猿类谱系基因表达多样性的了解。这些新信息大大提高了对基础生物学,物种多样性,进化,大脑发育以及本文强调的许多其他知识的认识,从而推动我们对自己“人之为人”的理解。可以说,这篇论文为现在和未来的比较基因组学领域的研究设定了一个新的标准,并且扩展到最终分析和比较个体人类之间的基因组的方式,并以实例说明了SMRT测序技术以及单分子光学图谱技术为这些类型的科学研究带来了有力的价值。
原文:High-resolution comparative analysis of great ape genomes
DOI号:10.1126/science.aar6343

来源:生物通
版权及免责声明:本网站所有文章除标明原创外,均来自网络。登载本文的目的为传播行业信息,内容仅供参考,如有侵权请联系答魔删除。文章版权归原作者及原出处所有。本网拥有对此声明的最终解释权。
{replyUser1} 回复 {replyUser2}:{content}